

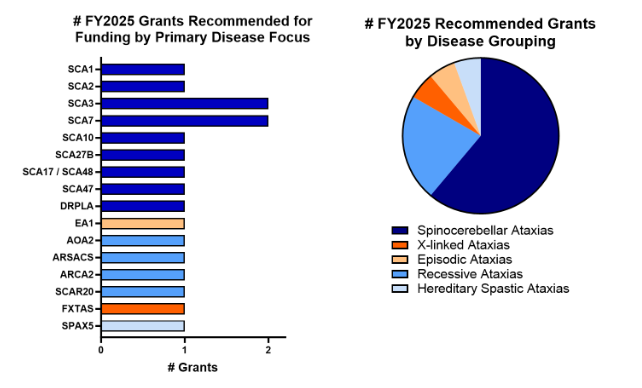
We’re proud to announce that we funded 18 research grants for 2025 totaling $730K!
For the funding term of March 1, 2025 – February 28, 2026, NAF received 96 Letters of Intent and 33 grant applications. ~50 scientific reviewers scored full applications, assigning a score between 1.0 and 5.0 (1= outstanding) for the following categories:
- Scientific Merit
- Innovation
- Impact
- Overall
Reviewers also provide written feedback on feasibility (budget & timeline), and alignment with NAF mission.
We’d like to give a huge kudos to our scientific reviewers who volunteer their time and expertise to make sure that the best science to accelerate Ataxia treatment development is funded!
For 2025, NAF funded the following research:
- 7 Research Seed Money Grants – totaling $350,000
- 3 Early Career Investigator Awards – totaling $150,000
- 5 Graduate Research Fellowships – totaling $125,000
- 3 Post-Doctoral Fellowship – totaling $105,000
To learn more about each type of research study, visit www.ataxia.org/researcher-resources.
A full list of the research studies and their lay summaries are available below.
Lay Summaries
Research Seed Money Grants
$50,000 – 1-Year Grant
48 LOIs Received | 13 Full Applications Reviewed | 7 Funded

Dr. Geoffrey Abbott
The Regents of the University of California, Irvine
Research Title: Small molecule therapeutic development for Episodic Ataxia 1
Ataxia Type: EA1
Episodic ataxias are debilitating movement disorders often also associated with a constellation of additional pathologies. We recently drew from Native American traditional medicine approaches to identify several plant metabolites that reverse the cellular effects of episodic ataxia-linked gene variants. In this pilot project, we will test the efficacy and safety of these promising compounds in a rodent model of human Episodic Ataxia Type 1, with the goal of preparing for future clinical trials. .

Dr. Naiara Aquizu Lopez
The Children’s Hospital of Philadelphia
Research Title: Uncovering a novel recessive spinocerebellar ataxia caused by genetic variants in SNX13
Ataxia Type: SCAR20
Disturbed lipid regulation is emerging as a major cause of recessive spinocerebellar ataxias, but mechanisms that lead to disease remain unknown. We recently uncovered a novel group of spinocerebellar ataxias caused by genetic mutations in putative lipid transport proteins. Capitalizing on these discoveries, our project aims to elucidate why the cerebellum is particularly vulnerable to lipid dysregulation and to establish the genetic diagnosis for a previously undefined group of recessive spinocerebellar ataxias.

Dr. Nicholas Brown
University of North Carolina at Chapel Hill
Research Title: Deciphering the link between mRNA regulation and protein degradation in PUM1-related ataxias, PADDAS and PRCA
Ataxia Type: SCA47
In spinocerebellar ataxia-47 (SCA47), mutations in the PUM1 gene can result in two distinct syndromes: Pumilio1-Associated Developmental Delay and Seizures (PADDAS) and Pumilio1-Related Cerebellar Ataxia (PRCA). To date, most studies have focused on how different mutations in PUM1 lead to varying phenotypes; but it is currently unclear how PUM1 is regulated. A deeper understanding of this process may be key to uncovering the physiological role of PUM1 in the brain..

Dr. Francois Gros-Louis
Laval University
Research Title: Generation of patient-derived Isogenic iPSC Lines of ARSACS Patients: A Critical Resource for Therapeutic Development
Ataxia Type: ARSACS
This project aims to advance the understanding and treatment of Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) by generating patient-derived induced pluripotent stem cells (iPSCs) and using CRISPR-Cas9 gene correction to create isogenic models. This initiative will provide valuable tools for the scientific community, accelerating drug discovery and fostering collaborative efforts to develop effective treatments for ARSACS and related genetic disorders.

Dr. Angela Laird
Macquarie University
Research Title: Understanding the role of cellular energetics and the gut-brain axis in MJD
Ataxia Type: SCA3
We have recently identified that the composition of the gut microbiome of a transgenic mouse model of SCA3/MJD is different to that of litter-mate control mice, even at ages prior to the onset of motor symptoms. Furthermore, we have identified that these mice develop altered gastrointestinal function. These changes have not been explored before. In this study, we will investigate the underlying causes of these changes and the effects of modifying underlying mechanisms.

Dr. Francesca Maltecca
San Raffaele Hospital
Research Title: Testing a gene therapy approach in the spastic ataxia type 5 (SPAX5) mouse model
Ataxia Type: SPAX5
Spastic ataxia type 5 (SPAX5) is a severe form of childhood-onset ataxia, associated with spasticity and epilepsy. It is caused by mutations in the AFG3L2 gene which encodes the homonymous protein AFG3L2, which resides in the mitochondrion and is essential for protein quality control, energy production and regulation of mitochondrial morphology. In agreement, we observed energy failure and altered mitochondrial morphology as key features SPAX5, demonstrated by our studies in cells derived from patients and in mouse models.
We recently analyzed cells derived from SPAX5 patients and we found that the levels of AFG3L2 protein are strikingly reduced. This disease represents therefore an ideal condition to test a gene replacement therapy based on adeno-associated viral (AAV) vectors. AAVs up to now represent the most promising and safest gene delivery tools for clinical trials, and have been successfully employed for severe neurodegenerative diseases. This encouraging data support a similar approach for treating SPAX5 with early intervention and a specific site of administration strategy, considering the seriousness and time of onset of this devastating recessive disease.
Based on promising preliminary data, the goal of our proposal is to investigate a gene-based therapeutic strategy to restore sufficient levels of AFG3L2 protein in the cerebellum and other neuronal systems, seeking to correct the ataxic phenotype and the mitochondrial dysfunction of the Afg3l2-/- mouse. The results obtained by our studies may open new perspectives for the treatment of SPAX5, for which a cure is still an unmet need.

Dr. Stephan Zuchner
Miller School of Medicine at the University of Miami
Research Title: Creating a rodent RGR14 GAA repeat expansion model as a translational platform for SCA27B
Ataxia Type: SCA27B
The goal of this project is to create a rat animal model that will mimic the pathogenic DNA changes seen in people with SCA27B. Such an animal model, once characterized, will represent a valuable platform to further study the disease pathology and also to safely and efficiently test new treatments for this form of ataxia. For that reason the animal will be ‘humanized’ at the SCA27B locus, containing human DNA stretches, which will allow the testing of genetic therapy approaches, such as ASO or CRISPR/Cas9.
Early Career Investigator Awards
$50,000 – 1-Year Grant
14 LOIs Received | 6 Full Applications Reviewed | 3 Funded

Dr. Joanna Korecka-Roet
Brigham and Women’s Hospital, Inc.
Research Title: Restoring disrupted DRPLA neuronal activity through genetic medicine
Ataxia Type: DRPLA
DRPLA is a rare, genetic, progressive brain disorder that causes ataxia, dementia and epilepsy, the latter particularly affecting young patients. Currently, no treatments are available. In this study, we aim to enhance our existing brain cell models that use stem cells from DRPLA patients to mimic the epilepsy-related changes ‘in a dish.’ We will also explore how astrocytes, the brain’s supportive cells, affect brain-cell activity in this disease. Using this model, we will test whether two new genetic therapies can correct the abnormal brain activity caused by this condition.

Dr. Tatsuaki Kurosaki
Jacobs School of Medicine and Biomedical Sciences
Research Title: RNA Misregulation in Spinocerebellar Ataxia Type 10 Cells
Ataxia Type: SCA10
Spinocerebellar ataxia type 10 (SCA10) is an inherited neurodegenerative disorder that causes progressive ataxia, seizures, cognitive impairment, and peripheral neuropathy. Preliminary transcriptome and proteome analyses suggest that SCA10 involves disruptions in RNA metabolism, misregulation of essential RNA-binding proteins, and interference with normal cellular functions. By studying multiple SCA10 cell models, we aim to further clarify the disease mechanism and contribute to the development of effective treatments.

Dr. Mafalda Raposo
i3S- Instituto de Investigação e Inovação em Saúde da Universidade do Porto
Research Title: Deciphering the role of long noncoding ATXN3 RNAs in Machado-Joseph disease/spinocerebellar ataxia type 3 (MJD/SCA3) pathogenesis
Ataxia Type: SCA3
Machado-Joseph Disease (MJD) also known as spinocerebellar ataxia type 3 (SCA3), is caused by a mutation in the ATXN3 gene. ATXN3 produces various forms of an RNA molecule known as alternative transcripts. Around 40% of these RNAs are expected to be long noncoding RNAs (lncRNAs). These lncRNAs do not create proteins but might help control gene expression. It’s still uncertain whether these lncRNAs have a protective role or contribute to MJD pathogenesis. Our recent research found three ATXN3 lncRNAs that may play a role in the disease process, but their precise function remains unclear.
This project will investigate several questions about these ATXN3 lncRNAs:
- Are their levels different in three brain areas and between healthy controls and MJD patients?
- Are they located in the nucleus, the cytoplasm, or both in the cells?
- Do they interact with DNA, RNA, or proteins?
- Is the normal function of these lncRNAs changed in the brain tissues of MJD patients?
By the end of this project, we will be able to determine whether ATXN3 lncRNAs vary in expression and interactions across brain regions with different levels of degeneration, potentially explaining why some regions are more vulnerable to the disease.
Post-Doctoral Fellowship Fellowship
$35,000 – 1-Year Grant
12 LOIs Received | 7 Full Applications Reviewed | 3 Funded

Dr. Luis Almaguer Mederos
University College London Institute of Neurology
Research Title: Investigating Genetic Modifiers of SCA2
Ataxia Type: SCA2
Spinocerebellar ataxia type 2 (SCA2) is a disease that affects the nervous system, and there are no preventive treatments. Our project seeks to identify genetic variants that explain the severity of disease manifestations using a Genome-Wide Association Study in a large international cohort of patients with SCA2. Identifying disease-modifying genetic variants will allow us to get deeper into the mechanisms of disease, recognize potential molecular markers of disease severity that might be useful to predict the course of disease and to stratify patients when testing potential therapies in clinical trials, improve genetic counseling in affected families, identify potential therapeutic targets and provide critical information for developing effective and targeted therapies to treat affected patients and at-risk family members.

Dr. Michael Almeida
University of North Carolina at Chapel Hill
Research Title: CHIP in Spinocerebellar Ataxias: Unraveling Cellular Distribution, Disease Mechanisms, and Neuroprotective Roles
Ataxia Type: SCA17/SCA48
Spinocerebellar ataxias (SCAs) are rare genetic disorders that impair coordination and movement due to cerebellar damage. This project investigates the role of CHIP, a protein involved in cellular protection, and how its mutations contribute to disease progression by disrupting its normal localization and function.
Using advanced imaging techniques, the study aims to uncover how CHIP’s dynamic distribution changes in healthy versus diseased brain cells, potentially leading to new therapeutic strategies for SCAs.

Dr. Débora Farina Gonçalves
Claude Bernard University Lyon 1
Research Title: Sulfide metabolism as a therapeutical target for autosomal recessive ataxia type 2 (ARCA2)
Ataxia Type: ARCA2
Autosomal recessive cerebellar ataxia type 2 (ARCA2), or COQ8A-ataxia, is a rare genetic disorder that leads to progressive symptoms such as balance difficulties, muscle weakness, seizures, and intellectual disability. This condition is caused by mutations in the COQ8A gene, which is crucial for producing Coenzyme Q10 (CoQ), a molecule necessary for energy production in cells and hydrogen sulfide (H2S) oxidation. A deficiency in CoQ damages brain cells, particularly Purkinje neurons (PN), which are vital for movement control. Our findings suggest that an early increase in hydrogen sulfide (H2S) may contribute to PN damage by disrupting energy production and causing mitochondrial dysfunction. Currently, no effective treatments are available for COQ8A-ataxia. Since CoQ is essential for the oxidation of H2S in cells, we aim to explore innovative ways to deliver CoQ to the brain using nanoparticles and develop strategies to reduce H2S toxicity using specific scavengers. This research aims to understand how H2S accumulation affects PN in COQ8A-ataxia and test potential treatments, which could benefit not only COQ8A-ataxia patients but also individuals with other CoQ deficiency-related diseases.
- COQ8A-ataxia is a rare genetic disorder, leading to symptoms like balance issues, muscle weakness, and seizures.
- This condition is caused by mutations in the COQ8A gene, which is crucial for producing Coenzyme Q10 (CoQ).
- COQ8A-ataxia damages particularly Purkinje neurons, by disrupting energy production.
- A toxic accumulation of hydrogen sulfide (H2S) at early disease stages may contribute to PN damage.
- Since oxidation of H2S is dependent of CoQ levels, we aim to improve CoQ delivery to the brain using nanoparticles and explore strategies to reduce H2S toxicity using specific scavengers.
- This work could offer potential treatments for COQ8A-ataxia and other CoQ deficiency-related diseases.
Graduate Research Fellowship
$25,000 – 1-Year Grant
17 LOIs Received | 7 Full Applications Reviewed | 5 Funded

Ms. Alison Chase
Yale University School of Medicine
Research Title: Identifying the astroglia-mediated pathogenic mechanisms contributing to neuronal vulnerability in Spinocerebellar Ataxia Type 1 (SCA1)
Ataxia Type: SCA1
Although Purkinje cell loss is the primary pathological hallmark of SCA1, other cell types, including glial “support” cells, express the mutant ATXN1 gene. While astroglia are the most abundant glial cell type in the brain, it remains unclear how they contribute to SCA1 pathology and progression. In this proposal, we have created a novel mouse model in which mutant ATXN1 is expressed selectively in astroglial populations. We plan to use this mouse model to uncover the molecular mechanisms by which astroglia affect the health of Purkinje cells and other neuronal subtypes in the context of SCA1. We hope this work will reveal new glial-specific targets that may be therapeutically beneficial in promoting neuronal health and preventing SCA1 disease progression.

Ms. Hailey Collis Napier
Duke University
Research Title: A Cross-Species Atlas of Purkinje Cell Subtypes to Identify Ataxia-Causing Genes
Ataxia Type: SCA7
I’m interested in Purkinje cells, a key brain cell type that is known to degenerate in Spinocerebellar Ataxia Type 7 (SCA7). My lab previously found that Purkinje cells in mouse models of SCA7 abnormally express hundreds of genes. The goals of my project are to better understand these dysregulated genes in SCA7, and to investigate how genes are regulated in Purkinje cells across many vertebrates. Similarities across species will help us to understand which findings in mouse models can be used to help human patients.

Ms. Judith Fishburn
The Regents of the University of California, Davis
Research Title: Dissecting the role of innate immune activation and reactive oxygen species in ataxia with oculomotor apraxia type 2 (AOA2)
Ataxia Type: AOA2
Our research aims to determine how DNA damage and reactive oxygen species (ROS) contribute to innate immune activation and oxidative damage in ataxia with oculomotor apraxia type 2 (AOA2) models and patient-derived cells. How these molecular phenotypes contribute to AOA2 disease phenotypes remains unknown, especially ROS given our lab recently identified this phenotype in an AOA2 mammalian model. Finally, this work will identify contributory pathways to disease etiology that are not well understood in AOA2, providing further insight into potential biomarkers for therapeutic targets.

Mr. Nolan Huck
The Regents of the University of California, Irvine
Research Title: Multiomic Investigation of SCA7 Retinal Degeneration: Linking Transcriptional and Epigenomic Dysregulation
Ataxia Type: SCA7
Spinocerebellar Ataxia type 7 (SCA7) causes progressive vision loss and impaired motor coordination. Unlike other ataxias, SCA7 uniquely affects the retina, leading to cone-rod dystrophy, a condition where patients lose color and low-light vision before progressing to complete blindness. This disease is caused by a mutation in the ATXN7 gene, which disrupts gene regulation in retinal cells, but the precise mechanisms driving retinal degeneration remain unclear. Our research will use single-cell and spatial technologies to investigate how SCA7 alters gene activity and DNA organization within the retina. By mapping these changes at the level of individual cells, we aim to pinpoint key molecular pathways driving retinal dysfunction. This work will provide new insights into SCA7 disease progression and help identify potential therapeutic targets to preserve vision in affected individuals.

Ms. Emily Timm
Rush University Medical Center
Research Title: The neural mechanisms underlying cortical control of gait and cognition in Fragile X-Associated Tremor/Ataxia Syndrome
Ataxia Type: FXTAS
Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS) is characterized by cognitive and cerebellar motor dysfunction causing mobility impairment, increased fall risk, and poor quality of life. Executive function deficits and dual tasking in FXTAS negatively impact gait, but a critical knowledge gap exists for neural mechanisms underlying these cognitive-motor relationships, which impedes development of rational treatment strategies. The fundamental goals of my project are to: 1) elucidate cortical activation patterns in FXTAS during walking and executive function tasks of varying difficulty using wireless functional near infrared spectroscopy (fNIRS) technology, 2) measure grey matter volume (GMV) in the prefrontal cortex, motor planning regions, and the cerebellum, white matter integrity (WMI) of frontal-cerebellar networks, and 3) associate GMV and WMI with cortical activation during simple and complex gait and executive function tasks, as well as spatiotemporal aspects of gait and executive function test scores.
View Past Years Funded Research
How Does the Research Grant Process Work?
To receive an Ataxia research grant from NAF, researchers go through a 4-step process:
- Submit a Letter of Intent (LOI) – This is a brief overview that gives information about the goal of their research study.
- LOIs are Reviewed – A panel of Ataxia experts review and score each LOI for scientific merit. They determine which LOIs will be invited to submit a grant application.
- Submit Grant Application – Studies which received a high enough score to move to the next step of the process are invited to submit a full application. The grant application requires more time and details about their project.
- Grants Applications Reviewed and Recommended for Funding – Each application receives 3 separate scientific reviews. NAF has 75 scientific reviewers who evaluate and score each full application that is received. Top-scoring grant applications are recommended for funding.
Thank you to all the researchers who submitted an LOI or application! Keep up the great work that is SO important to the Ataxia community!






